
Mesh surgical implants used to repair pelvic organ prolapse in women, a condition that frequently develops after childbirth, will face tougher federal scrutiny following thousands of injuries reported with the problem-prone devices.
The Food and Drug Administration said Monday that pelvic mesh will now be considered a class III, or high-risk, medical device, and manufacturers will be required to submit premarket approval applications demonstrating the safety and effectiveness of their products.
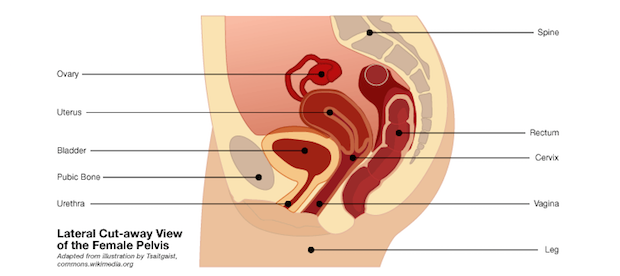
The change follows years of reports of pain, bleeding and infection among women who had the devices implanted to correct pelvic organ prolapse (POP). The condition occurs when when the bladder or other reproductive organs slip out of place, causing pain, constipation and urinary issues. The new FDA requirements do not apply to mesh products used to treat other conditions such as hernias or urinary incontinence.
Plastic mesh is often used to strengthen the pelvic wall and support internal organs in cases of prolapse. The mesh is often inserted through the vagina, using a small surgical incision. The Washington Post recently reported that as many as half of women may experience pelvic floor symptoms in their lifetime, and 200,000 undergo such operations each year.
The FDA action comes more than four years after the agency concluded that women getting vaginal mesh have more complications than women who undergo traditional surgery with stitches. Mesh products were introduced for pelvic repair in the 1990s and promoted as a way to speed patients' recovery time. But the FDA said in 2011 that about ten percent of women experienced complications from mesh, sometimes requiring multiple surgeries to reposition or remove it.
Patients have filed tens of thousands of lawsuits against mesh manufacturers, including Johnson & Johnson, Boston Scientific and Endo International. In 2014, Ireland-based Endo said it would pay $830 million to settle more than 20,000 personal injury lawsuits.
In a second rule, the FDA said vaginal mesh will now be classified as a "high-risk" medical device with a class III warning. Previously the implants were considered "moderate-risk" devices and carried a class II warning.
FDA recommends that women be aware of the risks associated with surgical mesh. On an advice page on the FDA website, the agency writes: "Ask your surgeon about all POP treatment options, including surgical repair with or without mesh and non-surgical options."
Non-surgical options include pelvic floor exercises known as Kegels. There are also non-invasive devices such as the PeriCoach, a smartphone-connected pelvic floor muscle training device for incontinence.
The FDA first proposed the changes announced Monday in 2014 draft orders.
Like 90 percent of medical devices sold in the U.S., pelvic mesh was originally cleared under a streamlined FDA review process for devices deemed similar to older products.